Archives
Fmoc-Leu-OH br Experimental Procedures br Author
Experimental Procedures
Author Contributions
Introduction
Frontotemporal dementia (FTD) refers to a group of neurodegenerative diseases caused by focal but progressive neuronal loss, astrogliosis, and spongiosis in the frontal and temporal cortices associated with abnormal intracellular accumulation of proteins, most commonly tau or TDP43 (TAR DNA-binding protein 43) (Karageorgiou and Miller, 2014; Neumann et al., 2015). Currently there are no effective disease-modifying therapies for FTD, making the treatment and prevention of FTD an area of significant unmet medical need.
Tau is expressed ubiquitously in the brain and locates predominantly in neuronal axons, where it regulates microtubule polymerization and guides the transport of proteins and organelles (Kosik et al., 1989; Morris et al., 2011). Alternative splicing of MAPT exons 2, 3, and 10 originates six tau isoforms that differ from one another by 29- or 58-amino-acid inserts at the N terminus, and by the presence of either three (3R-tau) or four (4R-tau) tandem-repeat sequences at the C terminus. Tau function and localization are regulated by post-translational modifications (PTMs); for example, phosphorylation, acetylation, and proteolysis (Johnson and Stoothoff, 2004; Min et al., 2010; Wang et al., 2009). In FTD, sporadic or autosomal dominant forms caused by MAPT mutations, inclusions containing hyperphosphorylated tau (P-tau) are detected within neurons and glia of affected brain regions.
Although these inclusions are key pathological features, the events leading to neuronal loss may start even earlier, but the tau species and precise molecular events causing cell death are poorly understood. Therefore it is crucial to investigate the early molecular events of disease, such as alterations in tau Fmoc-Leu-OH and affected cellular pathways (Gerson et al., 2014; Johnson and Stoothoff, 2004). In this context, human induced pluripotent stem cell (iPSC)-derived neurons allow exploring the molecular basis of tau pathogenesis in a disease-relevant genetic background (Ehrlich et al., 2015; Haggarty et al., 2016; Iovino et al., 2015).
Here, we investigated the underlying molecular and cellular mechanisms of pathogenicity associated with the rare tau variant A152T in a human neuronal context. Although the role of tau A152T in disease is still debated, it has been shown to affect tau function and PTMs, promote oligomerization and postmortem detection of inclusions, cause neuronal dysfunction independent of aggregation and neuroinflammation in animal models, and increase significantly the risk for FTD and other neurodegenerative diseases (Coppola et al., 2012; Kara et al., 2012; Labbe et al., 2015; Lee et al., 2013; Maeda et al., 2016; Decker et al., 2016; Pir et al., 2016; Sydow et al., 2016). We utilized iPSCs derived from A152T carriers and derived neural progenitor cells (NPCs) and differentiated neuronal cells (Figure 1A). These cells represent ex vivo models of human neurons, with tau expression at endogenous physiologically relevant levels and in the context of the genomic background associated with disease.  Overall, our results reveal potential targets for disease-modifying therapeutics to affect FTD and other tauopathies.
Overall, our results reveal potential targets for disease-modifying therapeutics to affect FTD and other tauopathies.
Results
Discussion
Human iPSC-derived neurons allow measurement of disease-relevant cellular and molecular phenotypes in a physiologically and genomically relevant context, potentially recapitulating the early stages of disease etiology,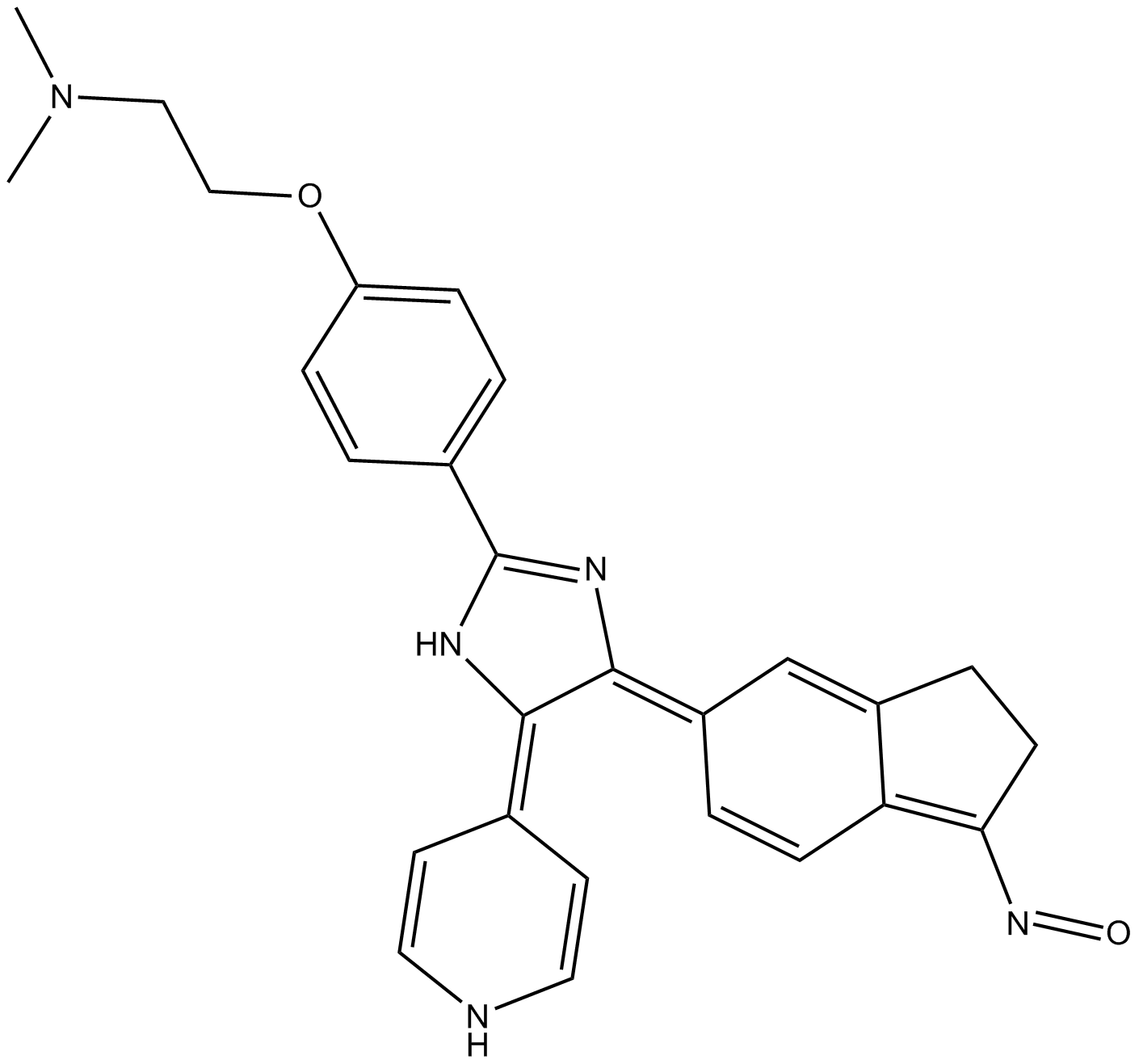 and allow direct testing of therapeutic targets and small molecules in a human neuronal environment (Almeida et al., 2012; Bilican et al., 2012; Ehrlich et al., 2015; Fong et al., 2013; Haggarty et al., 2016; Iovino et al., 2015; Wren et al., 2015). We took advantage of this system to investigate the early events of tau-A152T pathology that may be causal in FTD. Our findings contribute to mounting evidence, across studies of different tau mutations and across model systems, of tau-mediated molecular events associated with neurodegeneration, toward the identification of relevant therapeutic targets (Ehrlich et al., 2015; Fong et al., 2013; Iovino et al., 2015; Maeda et al., 2016; Decker et al., 2016; Pir et al., 2016; Sydow et al., 2016; Wren et al., 2015). We biochemically profiled neurons derived from control and A152T carriers, and report on a phenotyping platform for tau regarding protein levels, PTMs, and solubility. We demonstrate accumulation of endogenous tau/P-tau in A152T human neurons by multiple powerful methods. By western blot we found a significant upregulation of P-tau in all A152T neurons, whereas total tau upregulation was specific to the FTD19-derived neurons, consistent with previous reports (Fong et al., 2013; Sydow et al., 2016). This result might be a consequence of variability among individuals of the same MAPT genotype, and possibly the clinical outcome, but only future analysis of larger cohorts of cases could allow us to draw accurate conclusions. By MS analysis we verified upregulation of absolute levels of endogenous tau in A152T neurons that became accentuated with time. By targeted MS, we demonstrated differential levels of tau A152T versus the non-mutant form in A152T neurons, which is relevant to the identification of “the toxic species.” Even though this ratio (∼60% versus 40%, respectively) was constant during the period of time tested (Figure 4C), the gradual increase in total tau (Figure 4B), suggests that this amount of tau-A152T is sufficient to cause some imbalance that leads to overall accumulation of tau.
and allow direct testing of therapeutic targets and small molecules in a human neuronal environment (Almeida et al., 2012; Bilican et al., 2012; Ehrlich et al., 2015; Fong et al., 2013; Haggarty et al., 2016; Iovino et al., 2015; Wren et al., 2015). We took advantage of this system to investigate the early events of tau-A152T pathology that may be causal in FTD. Our findings contribute to mounting evidence, across studies of different tau mutations and across model systems, of tau-mediated molecular events associated with neurodegeneration, toward the identification of relevant therapeutic targets (Ehrlich et al., 2015; Fong et al., 2013; Iovino et al., 2015; Maeda et al., 2016; Decker et al., 2016; Pir et al., 2016; Sydow et al., 2016; Wren et al., 2015). We biochemically profiled neurons derived from control and A152T carriers, and report on a phenotyping platform for tau regarding protein levels, PTMs, and solubility. We demonstrate accumulation of endogenous tau/P-tau in A152T human neurons by multiple powerful methods. By western blot we found a significant upregulation of P-tau in all A152T neurons, whereas total tau upregulation was specific to the FTD19-derived neurons, consistent with previous reports (Fong et al., 2013; Sydow et al., 2016). This result might be a consequence of variability among individuals of the same MAPT genotype, and possibly the clinical outcome, but only future analysis of larger cohorts of cases could allow us to draw accurate conclusions. By MS analysis we verified upregulation of absolute levels of endogenous tau in A152T neurons that became accentuated with time. By targeted MS, we demonstrated differential levels of tau A152T versus the non-mutant form in A152T neurons, which is relevant to the identification of “the toxic species.” Even though this ratio (∼60% versus 40%, respectively) was constant during the period of time tested (Figure 4C), the gradual increase in total tau (Figure 4B), suggests that this amount of tau-A152T is sufficient to cause some imbalance that leads to overall accumulation of tau.